DANGLE takes only one input file, which contains the primary sequence and the chemical shift table.

All information should be put in-between a pair of tags:
| <seq_1>...</seq_1> | Mandatory | The primary sequence of the query protein in single letter code; can contain white spaces, be in lower or upper case, or use "X" to represent unknown residues. |
| <res_0>...</res_0> | Mandatory | The residue number of the first amino acid in the primary sequence given in the "seq_1" fields; must be ≥ 0. |
| <cs_data>...</cs_data> | Optional | Chemical shift data for the query protein; each line should contain the residue, residue code, atom_name and chemical shift value. Residue code can be in 3- or 1-letter-code. Chemical shift values are expected to be properly referenced. |
| <-!...-> | Optional | Comment field; not read by the program. |
You can add other information for book-keeping purposes, DANGLE does not read it.
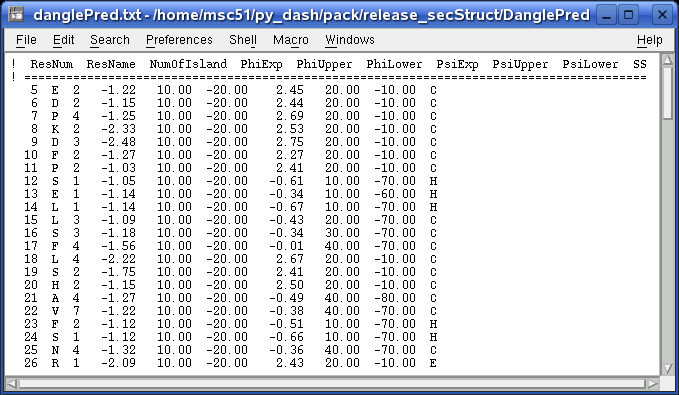
This file contains all the prediction information in columns: num of islands in GLE diagram, φ and ψ predictions, upper and lower limits, and secondary structure assignments.
Each residue has its own GLE diagram, unless it has been rejected (i.e. having more than a user-defined number of islands). The GLE diagram is created in the black-and-white PGM format. It can be converted to a coloured PGM diagram with a given script or other software.
| PGM | PPM |
 |
 |
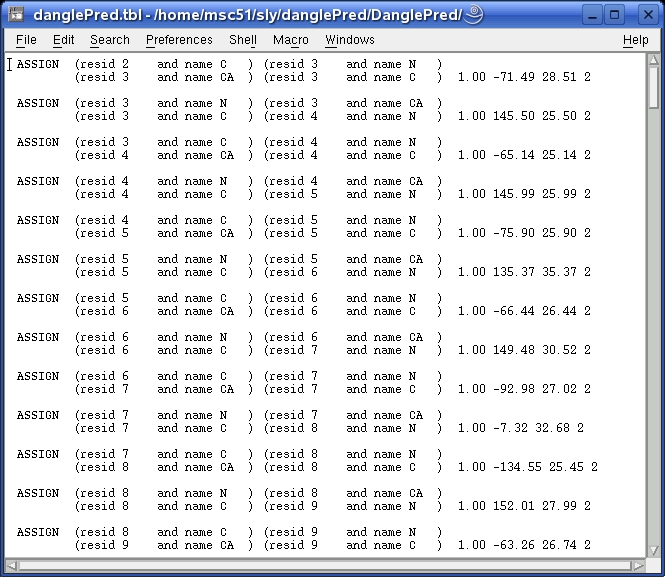
This .tbl file defines the φ and ψ constraints predicted by DANGLE and can be used directly in the ARIA program for structure calculation.
Copyright (C) 2009 Nicole Cheung, Tim Stevens, Bill Broadhurst (University of Cambridge)