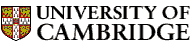
When no rejection is allowed, DANGLE makes prediction for every residue except the flanking two of the sequence. The number of distinct islands in a GLE diagram reflects the degeneracy of mapping between the secondary shift set of a query fragment and angle predictions in Ramachandran space. We recommend to treat GLE diagrams that contain many islands with caution. Systematic exclusion of residues that yield more than a set number of islands greatly improves the accuracy of the remaining predictions.
DANGLE allows user to specify a rejection level in terms of the number of islands in a GLE diagram, above which the program avoids making a prediction rather than to suggest a potentially wrong restraint.
Here are some examples showing how GLE diagrams look like for residues in different regions:
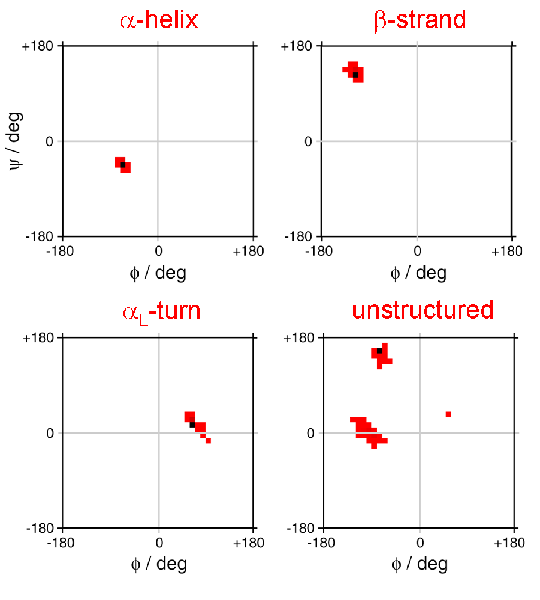
The performance of DANGLE was evaluated by analysing the sequence and chemical shifts of 27014 residues from the fragment database.
The following plots show the percentage of angle predictions made by DANGLE that were accurate to within ±30° for φ and ψ, for different residue classes, before and after omitting GLE diagrams that contain more than a set number of islands.
The improvement is most obvious when accuracy was defined as both φ and ψ being within ±30° of the reference angle. For example, when GLE diagrams that contain more than one island were discarded, 88% of the remaining predictions were accurate to within 30° of the reference for both φ and ψ, compared with 80% when all predictions were considered. Particularly notable is the improvement for glycine residues: the joint φ and ψ accuracy increased from 59% to 76%. However, the cost of filtering out multi-island predictions is that 18% of all estimations were rejected, comprising 15% of generic residues, 41% of glycines, 22% of prolines and 24% of pre-proline sites.
A different way forward would be to recognize that a multi-island GLE diagram presents information about possible alternative conformations, making it reasonable to consider the use of ambiguous dihedral angle restraints. Predictions could then be defined as being accurate if both reference angles appeared within the boundary ranges of any of the islands in the relevant GLE diagram, not just the principal cluster. In this case, >90% if all φ and ψ estimates would be classified as accurate, irrespective of the level of filtering.
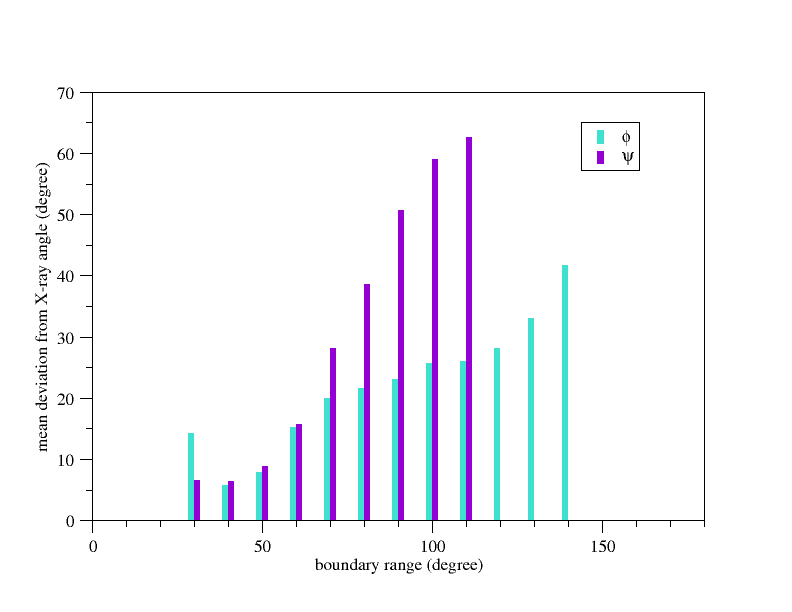
A clear correlation between the mean deviations of predicted angles from the reference structrue and the boundary range selected for φ and ψ means that a broad range indicates a realistic degree of uncertainty in the estimate. When all multi-island estimates were rejected, 95% and 93% of reference φ and ψ angles, respectively, were found within the boundary ranges suggested by DANGLE.
The following results were obtained by testing different prediction methods on an independent set of 29 proteins having crystal structures with resolution < 3 Å, varied in tertiary structures and with nearly complete shift sets. PREDITOR was tested in two modes: PRED-A, using only sequence and shift data; and PRED-B, supplementing the sequence and shifts with information from homologous structures.
The table shows the percentage of correct predictions, where a prediction was defined as accurate if, for both φ and ψ, the deviation from the experimental angle is within 30°.
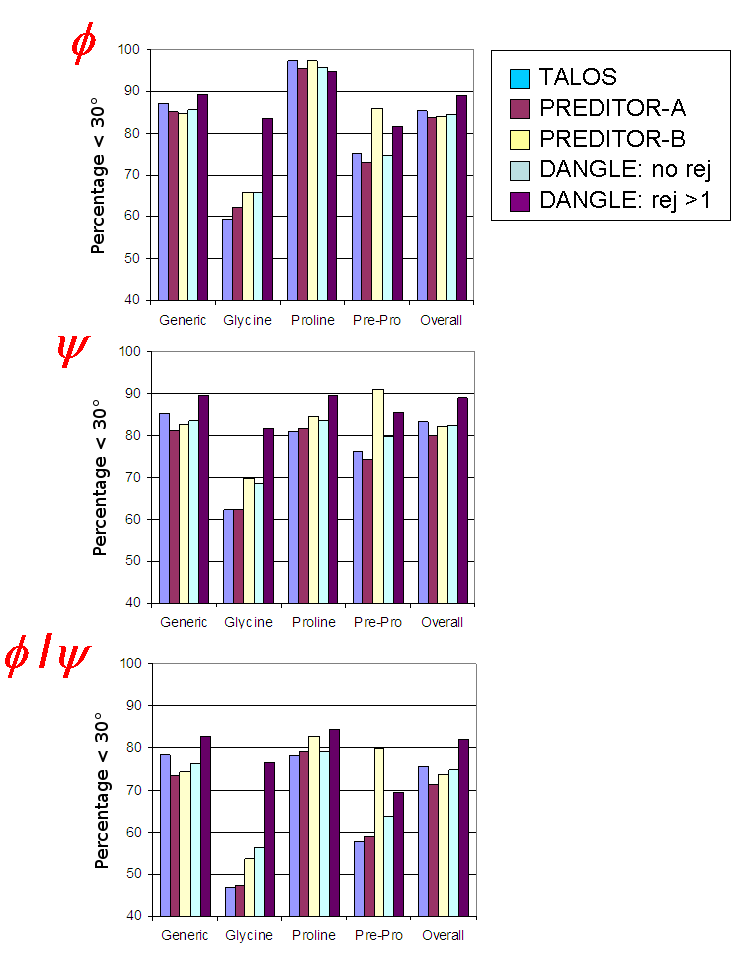
In the absence of filtering, the overall performance of DANGLE was marginally worse than TALOS and slightly better than PREDITOR. Treating the four amino acid classes separately, the accuracy rate for DANGLE was increased by 10% and 6% from TALOS for glycine and pre-proline residues, respectively. The accuracy of pre-proline predictions by PRED-B exceeded those of all other methods, suggesting that chemical shift information alone may not be sufficient to characterize the ψ angles at these sites.
When estimates from all ambiguous cases were discarded (17%), the remaining φ and ψ predictions were significantly more accurate than those from the other approaches, yielding an accuracy rate of 82 % for DANGLE, compared with 76 %, 71 % and 74 % for TALOS, PRED-A and PRED-B, respectively. The overall RMS error for DANGLE decreased from (φ=30.7°, ψ=45.8°) when all predictions were retained to (22.1°, 31.4°) when multi-island GLE diagrams were rejected, proving that filtering made the method more reliable than PRED-B, which gave RMS errors of (33.2°,38.5°), TALOS with (33.7°,50.7°) and PRED-A with (36.7°51.6°).
TALOS: Cornilescu G, Delaglio F, Bax A. J. Biomol. NMR. 1999; 13: 289-302.
PREDITOR: Berjanskii MV, Neal S, Wishart DS. Nucleic Acids Res. 2006; 34:W63-9
Copyright (C) 2009 Nicole Cheung, Tim Stevens, Bill Broadhurst (University of Cambridge)